History: Immediately following delivery to a healthy young woman, a baby boy was intubated for respiratory distress. An echocardiogram was performed on the child, revealing left ventricular enlargement with multiple ventricular masses that partially obstructed the outflow tract. Given the severity of his condition and the extent and location of the masses, the boy was deemed to be a poor surgical candidate. Ventilatory support was withdrawn and the boy expired shortly thereafter.
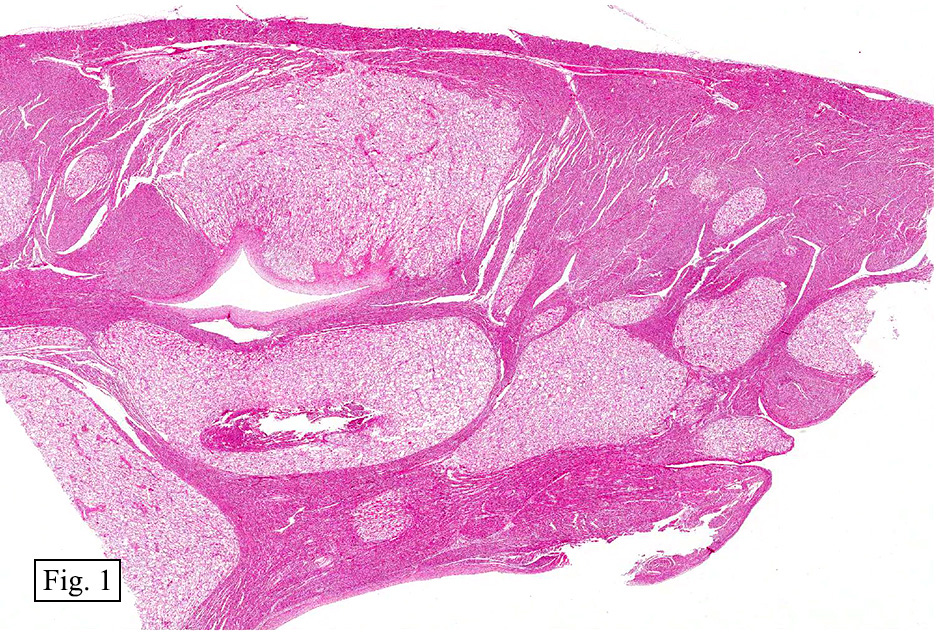
Gross examination of the heart at autopsy revealed numerous firm, white-tan ventricular mural nodules up to 1.8 cm. Although unencapsulated and apparently well-circumscribed, the nodules involved a substantial portion of the myocardium with some protruding into the ventricular cavity and approximating the mitral and aortic valves.
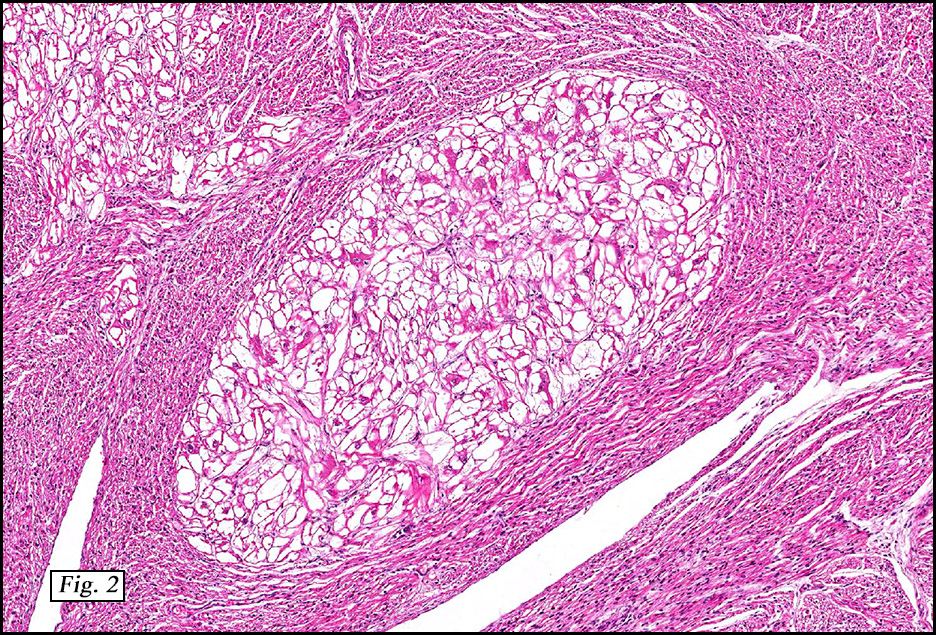
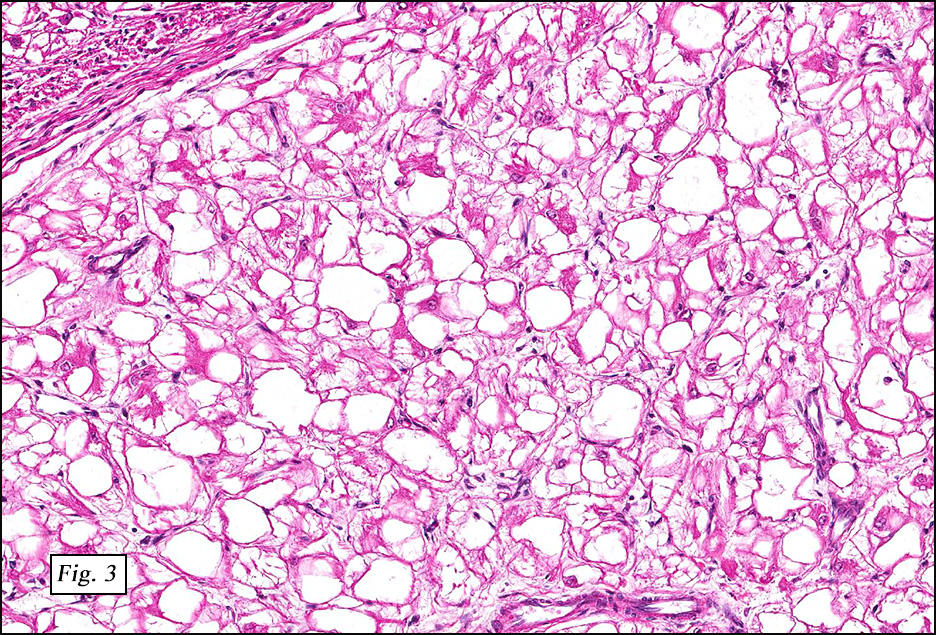
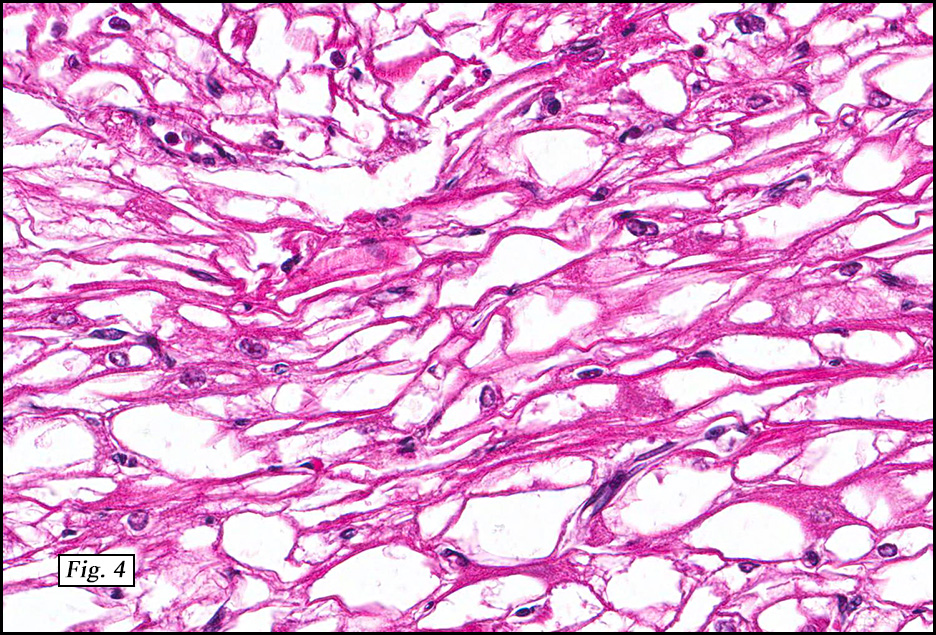
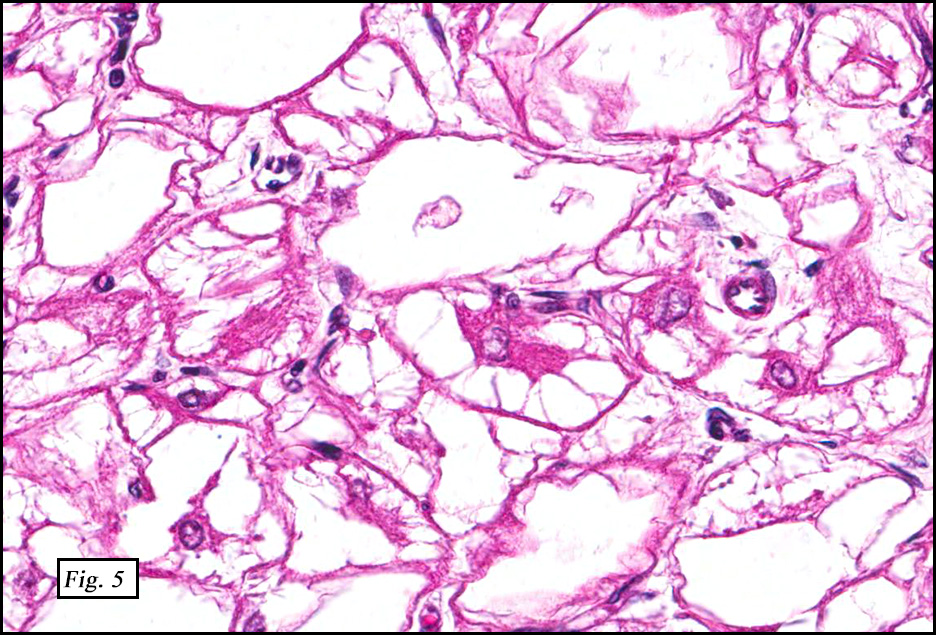
Microscopically, the tumors were comprised of large, polygonal clear cells which were distinct from the surrounding normal myocardiocytes (Figs. 1, 2, 3). The lesional cells were distended by cytoplasmic vacuoles which surrounded the nuclei (Fig. 3 & 4). Delicate wisps of pink cytoplasm emanated from the small, central nuclei extending to the periphery (Fig. 5). Significant atypia, mitotic figures, and necrosis were not encountered.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagnosis: Cardiac Rhabdomyoma
Elif L. Akin, M.D. and Donald R. Chase, M. D.
Department of Pathology & Human Anatomy
Loma Linda University Medical center, Loma Linda, California
Discussion: Cardiac rhabdomyoma (CR) is the most common cardiac neoplasm in the pediatric population, comprising about 60% of cardiac tumors in pediatric autopsy series and an overwhelming 90% of cardiac neoplasms diagnosed in utero. Considered to be a benign hamartoma which has no predilection for either gender, this neoplasm most frequently arises in the ventricles.
Grossly, cardiac rhabdomyomas are distinct, well-circumscribed white-tan nodules which contrast with surrounding myocardium. They can occur as solitary or multiple lesions, ranging in size from a few millimeters up to 10 cm. Although they usually arise in the left ventricular free wall, they can involve the muscle surrounding any of the cardiac chambers.
Microscopically, CR are characterized by nodules of round or polygonal clear cells that are much larger than myocardiocytes and swollen with glycogen-rich vacuoles. Nuclei are small and centrally located and have delicate connections to the perimeter of the cell comprised of eosinophilic wisps of cytoplasm, which are thought to represent invaginations of the cytoplasmic membrane. These so-called “spider cells†are characteristic of cardiac rhabdomyomas. Their peripherally vacuolated, clear appearance is a result of glycogen deposition, which is lost during routine processing. Striations may also be seen. While some CR may show degenerative change, significant nuclear pleomorphism, mitotic activity, and necrosis are not typically present.
Many CR occur as asymptomatic lesions that eventually spontaneously regress. As the regression rate is an estimated 50 to 70%, surgical intervention is often unnecessary, making CR the second most common cardiac tumor in pediatric surgical series (behind cardiac fibroma). When symptomatic, the clinical presentation of children with CR varies considerably depending on the size, extent, and location of the tumor(s). It is not altogether surprising that lesions involving the conduction system may cause arrhythmias and that large CR projecting into the ventricular cavity may cause flow abnormalities and mechanical obstruction. Potential clinical manifestations of problematic CR include murmurs, arrhythmias, and heart failure and may necessitate partial surgical resection, depending on symptom severity.
Although CR can occur as a solitary, isolated lesion, it is most frequently seen in the context of tuberous sclerosis complex (TSC). When associated with TSC, lesions tend to be multifocal and extend into the ventricular cavity. Because of the strong link to TSC, patients with a diagnosis of CR must be assessed for other signs of this disease. Cardiac rhabdomyomas are less often associated with congenital cardiac malformations including tetralogy of Fallot, Ebstein anomaly, and hypoplastic left heart syndrome.
There are several entities (including non-neoplastic lesions) which may be considered in the differential diagnosis of cardiac rhabdomyoma.
• Glycogen storage disease (Pompe disease) is also characterized by vacuolated myocytes. However, the cytoplasmic vacuoles tend to be central, with myofibrils at the periphery. In contrast to CR, glycogen storage disease does not form distinct nodules and lacks spider cells.
• Purkinje cell hamartoma (histiocytoid cardiomyopathy) is a rare pediatric cardiac tumor which also presents as multiple intramural nodules. Although this condition also has a predilection for the left ventricle, the nodules tend to be subendocardial. Unlike CR, the cells of histiocytoid cardiomyopathy (as the name would suggest) resemble histiocytes, are quite a bit smaller than spider cells, and show finely vacuolated cytoplasm filled with mitochondria, rather than glycogen.
• As the most commonly excised pediatric cardiac tumor, the shear frequency of cardiac fibroma makes it another important consideration in the differential diagnosis. While CR may be multifocal, cardiac fibromas are typically solitary mural lesions which resemble fibromatoses. Characterized by bland spindled cells interspersed with collagen bundles, fibromas tend to have infiltrative borders. Additionally, cardiac fibromas may demonstrate focal calcification, a feature not ordinarily observed in CR.
• Lipomatous hypertrophy, in contrast to the above conditions, affects older, usually obese adults. It preferentially involves the atrial septum and is distinguished from CR by the presence of three cell populations including brown and white adipocytes as well as hypertrophic myocytes.
In summary, CR is a benign hamartomatous neoplasm of infancy which typically arises in the left ventricular wall. It is characterized by spider cells: large cells with cytoplasmic vacuolization and central nuclei with strands of eosinophilic cytoplasm extending to the cell borders. While these lesions are usually asymptomatic and spontaneously regress, they can cause serious clinical problems (e.g., heart failure) and may require surgical intervention. Appropriate recognition and diagnosis of CR is important to determine whether surgery is required and to initiate further evaluation for possible tuberous sclerosis complex.
Suggested Reading:
Burke A, Tavora F. Practical Cardiovascular Pathology. Wolters Kluwer Health Lippincott Williams & Wilkins. 2011: 375-380.
Stocker JT, Dehner LP, Husain AN. Stocker & Dehner’s Pediatric Pathology: 3rd ed. Wolters Kluwer Health Lippincott Williams & Wilkins. 2010: 566-569.
Virmani R, Burke A, Farb A. Atlas of Cardiovascular Pathology. W. B. Saunders Company. 1996: 82-91.
Weiss SW, Goldblum JR. Enzinger & Weiss’s Soft Tissue Tumors: 5th ed. Mosby Elsevier. 2008: 583-592.