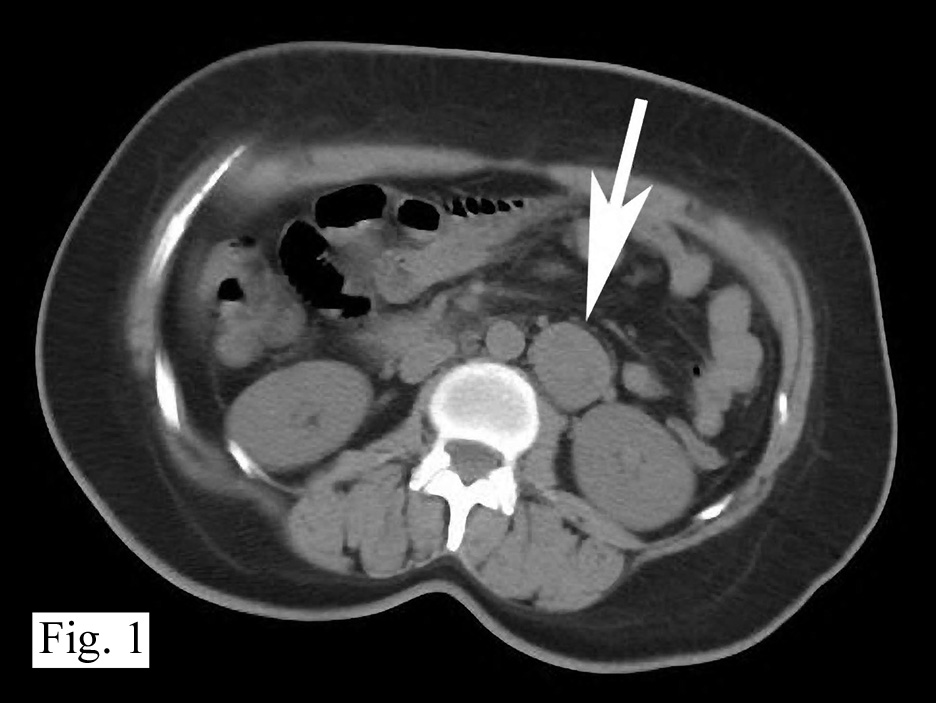
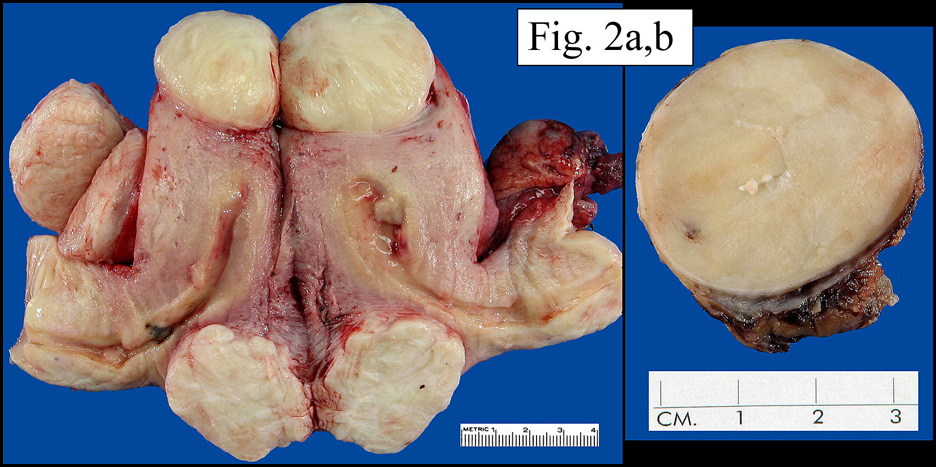
 History: A 32 year old previously healthy Hispanic male presented with a 3-week history of dyspnea upon exertion. Â A CT-scan showed a mass in the left cardiac atrium which extended into the pulmonary veins. Â The tumor was well encapsulated and attached to an atrial appendage. Â It measured 6 x 5 x 4 cm and was solid, pale pink-tan with gelatinous regions. Â Foci of necrosis and hemorrhage were present. Â The surgeon was not able to completely excise the mass.
History: A 32 year old previously healthy Hispanic male presented with a 3-week history of dyspnea upon exertion. Â A CT-scan showed a mass in the left cardiac atrium which extended into the pulmonary veins. Â The tumor was well encapsulated and attached to an atrial appendage. Â It measured 6 x 5 x 4 cm and was solid, pale pink-tan with gelatinous regions. Â Foci of necrosis and hemorrhage were present. Â The surgeon was not able to completely excise the mass.
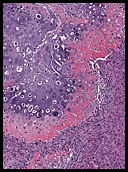
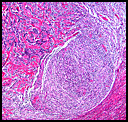
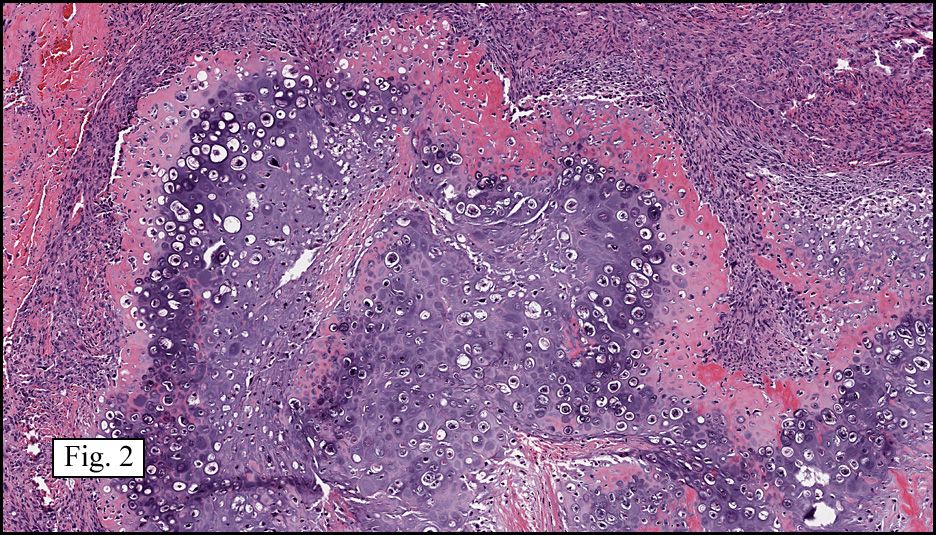
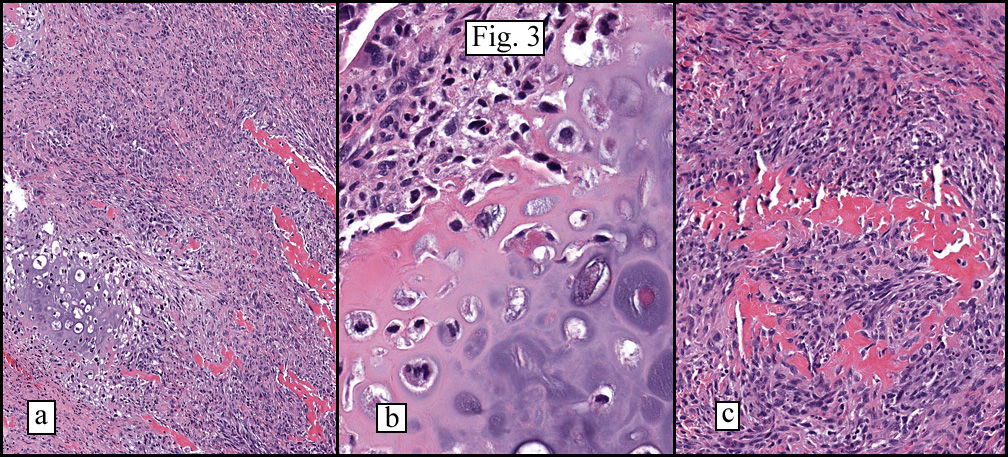
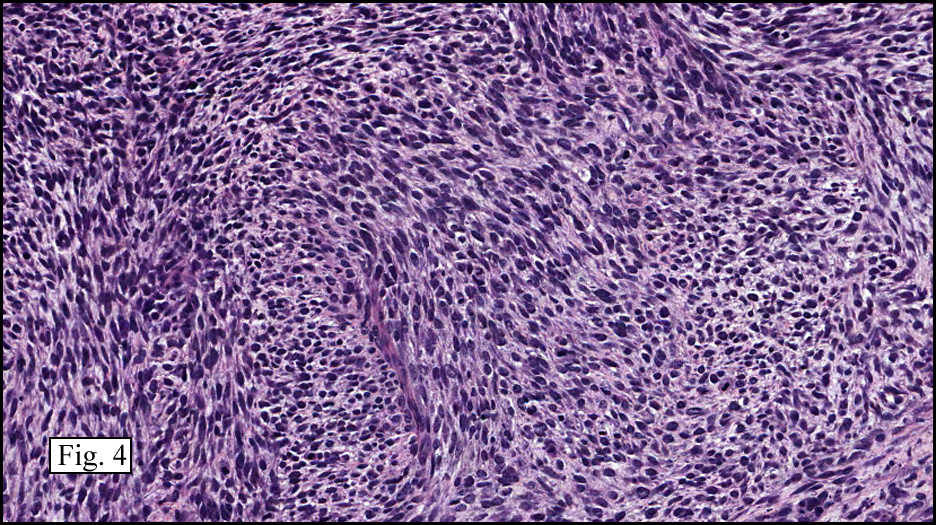
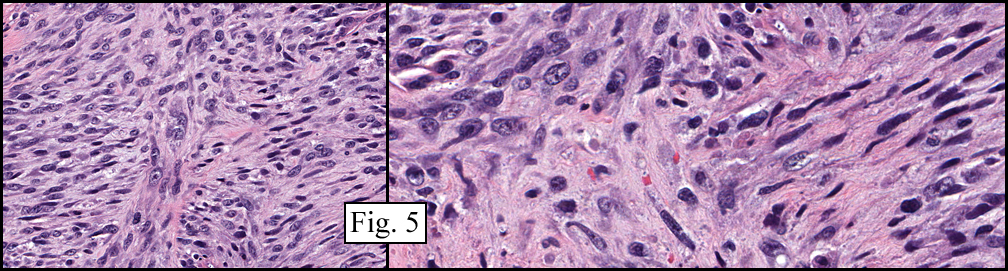
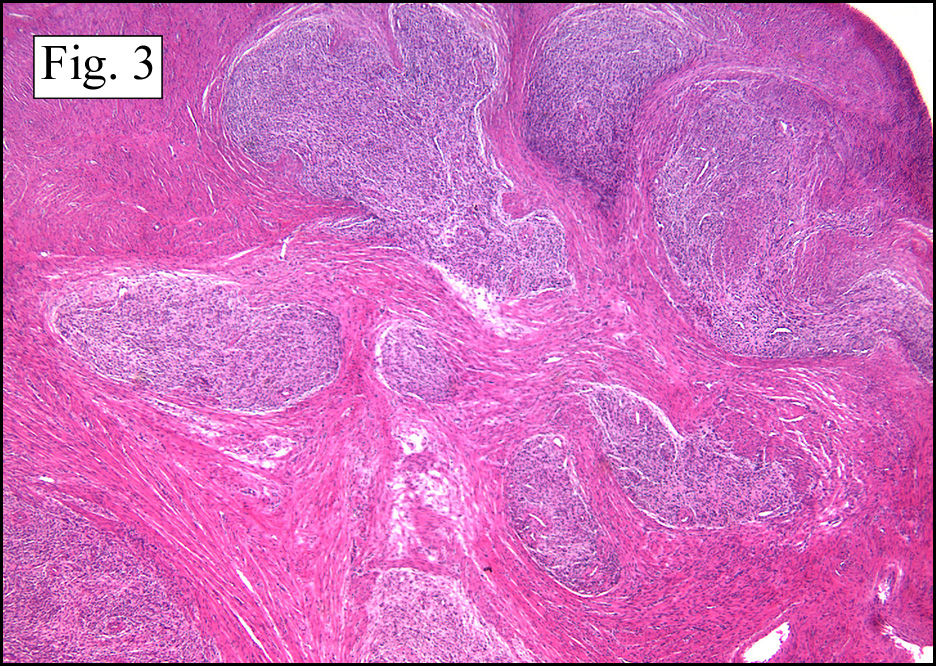
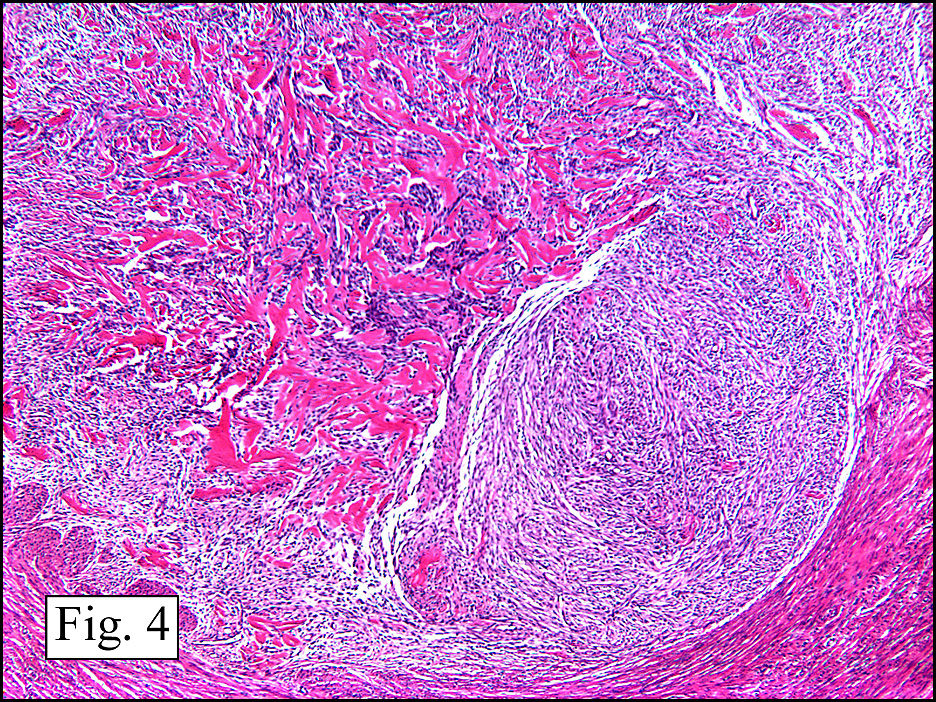
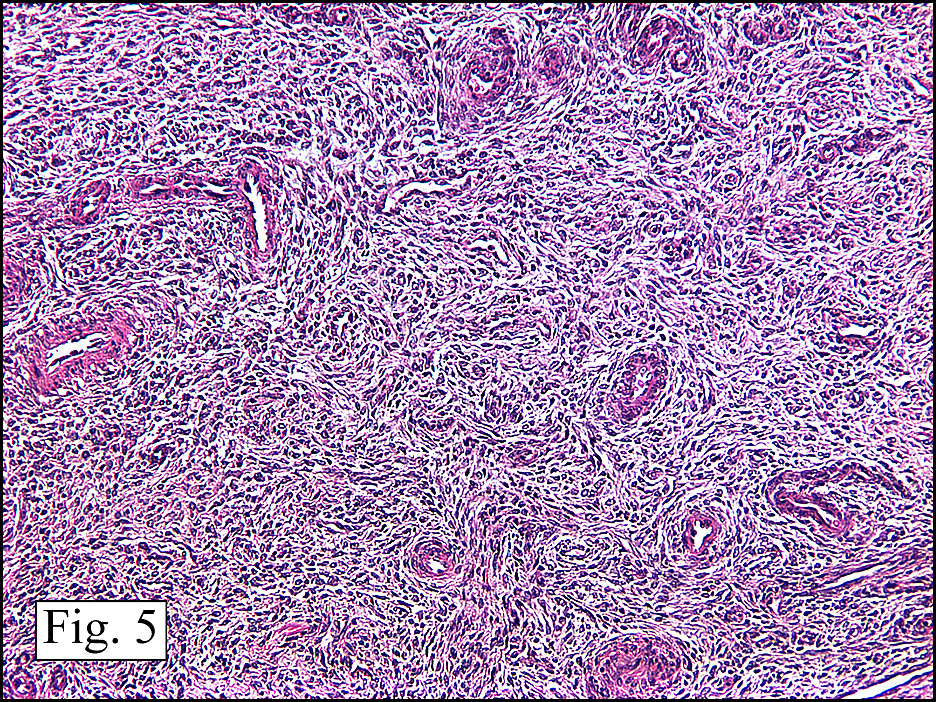
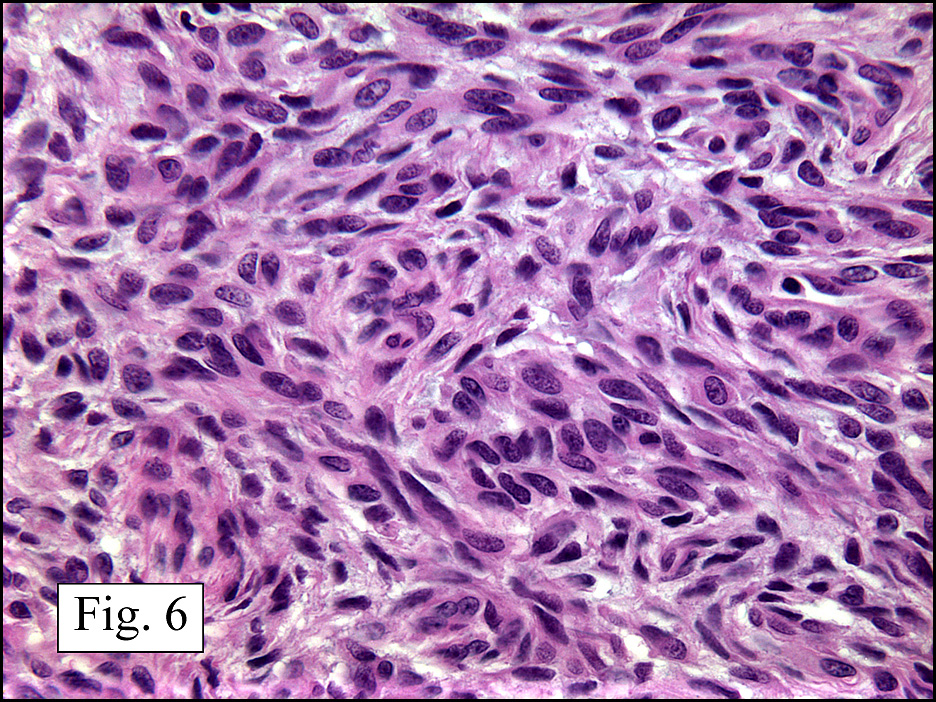
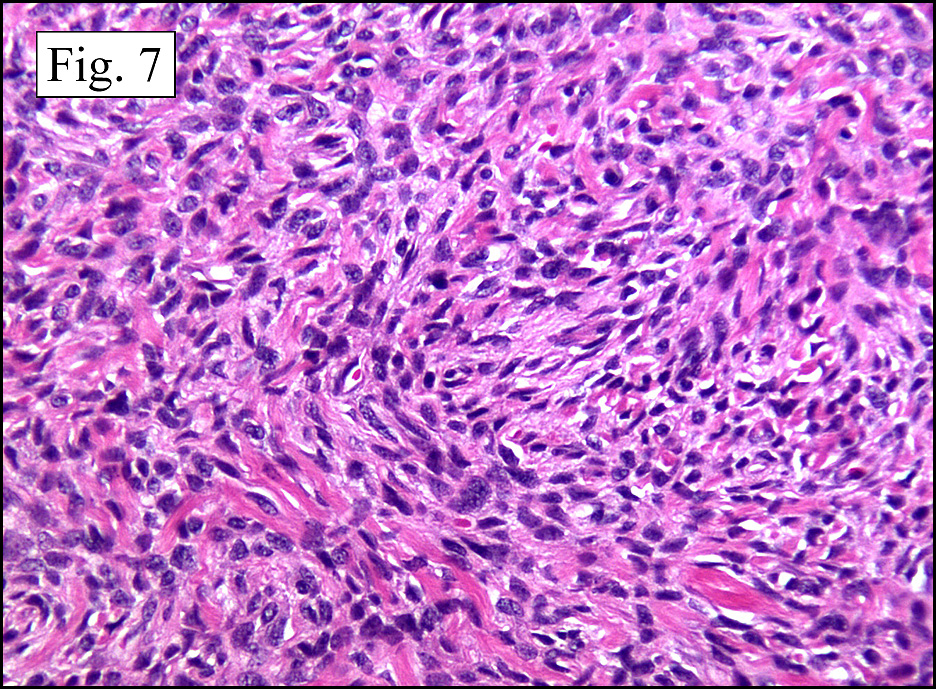
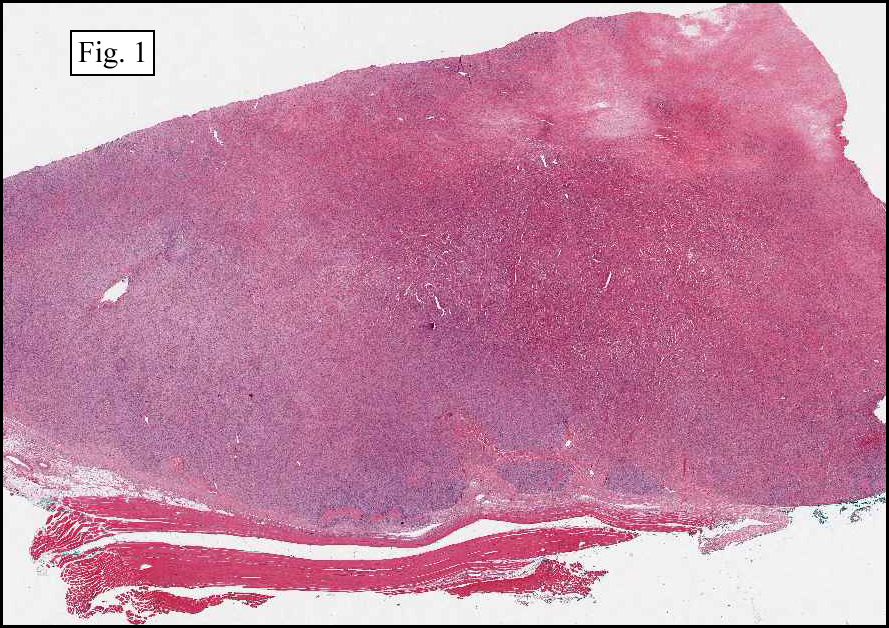
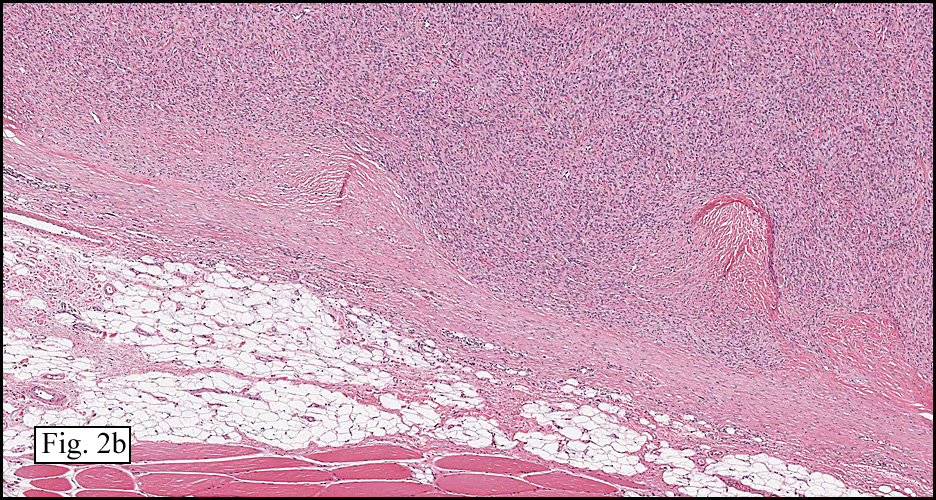
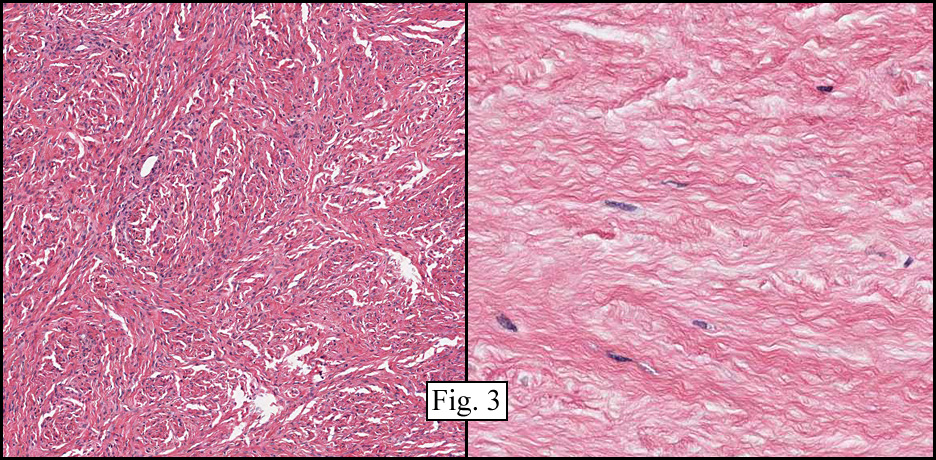
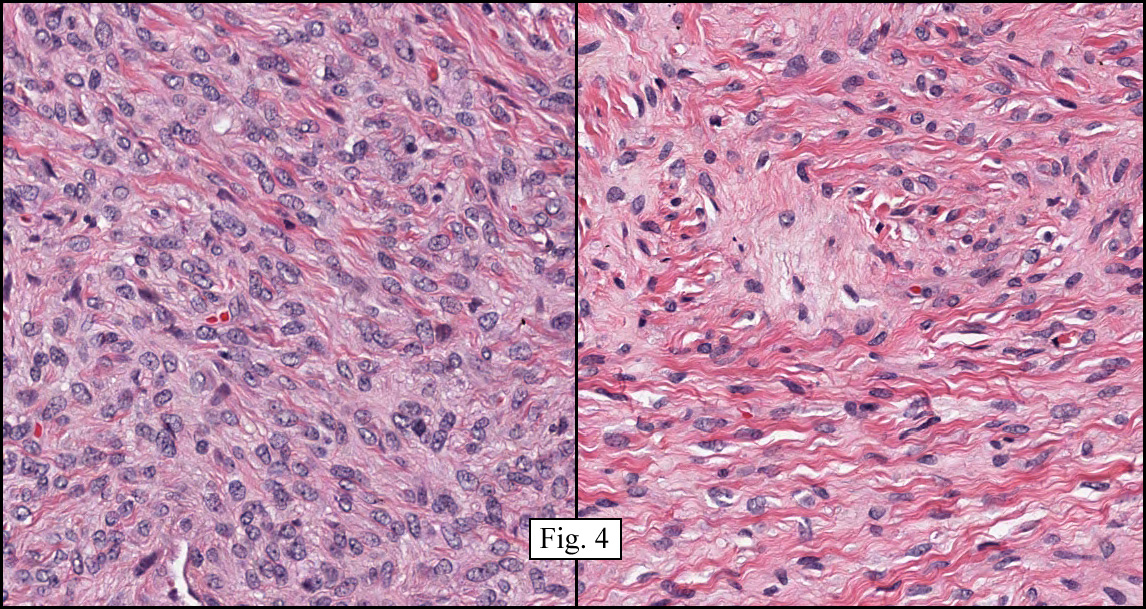
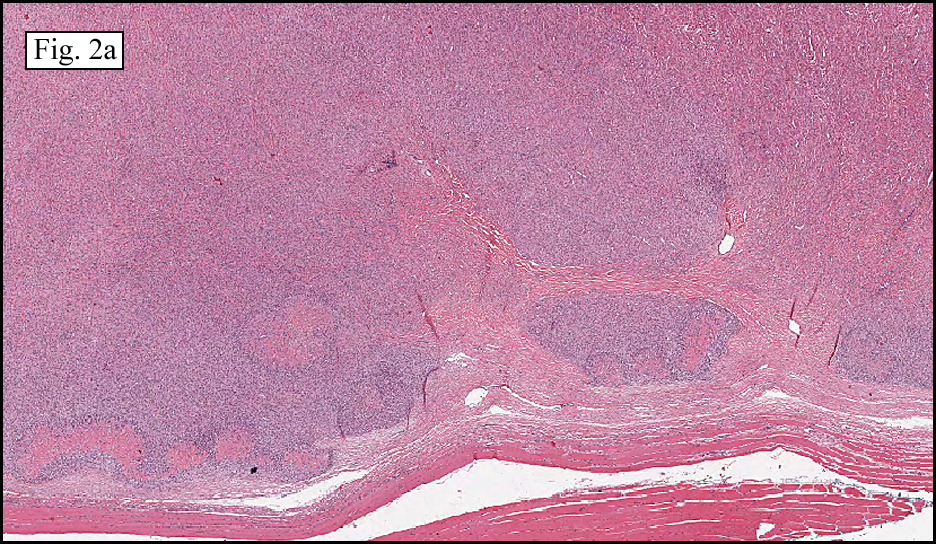
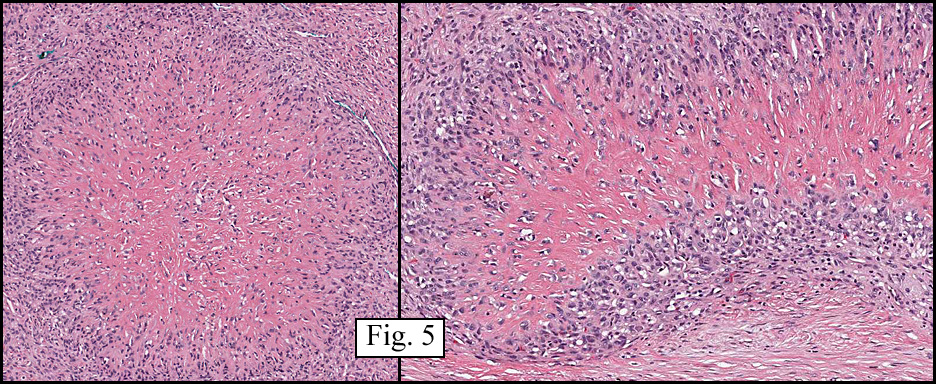
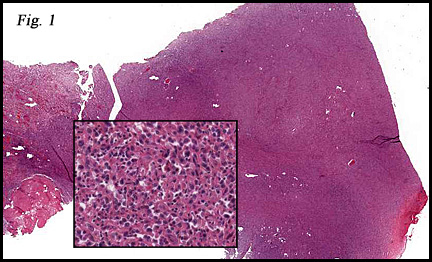
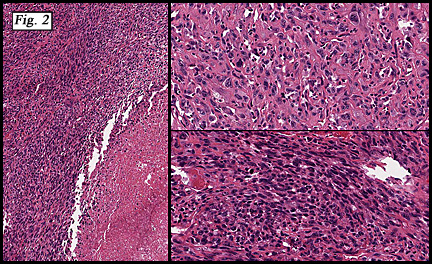
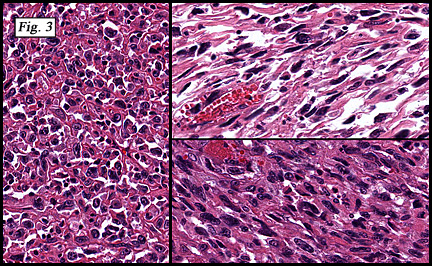
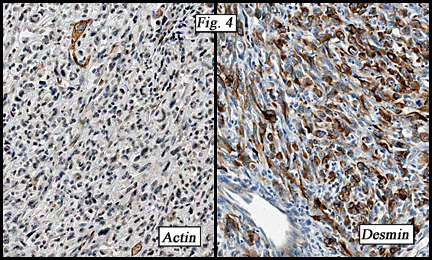
The neoplasm was microscopically remarkable for having a large number of sarcoma patterns (Fig. 1). Â There was a blending of spindle-cell sarcoma with osteosarcoma and chondrosarcoma (Fig. 2,3) Other areas consisted of sheets of uniform fusiform cells growing in long sweeping fascicles with a faint herringbone pattern (Fig. 4). Â Yet other regions showed cells with more pleomorphism and abundant eosinophilic cytoplasm with faint longitudinal and cross striations (Fig. 5).
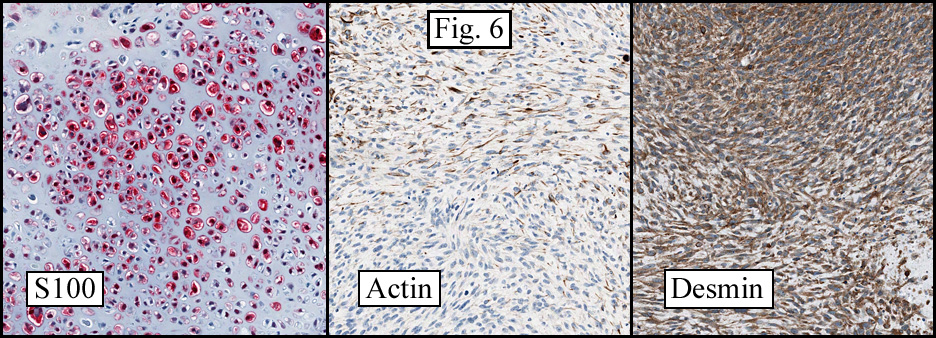
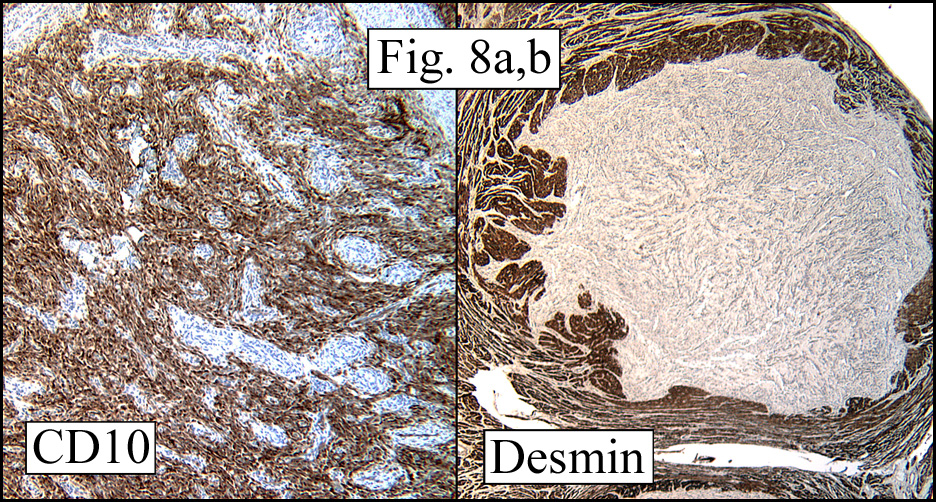
The cartilaginous, but not the spindle cell areas marked for S100 protein. An actin stain was weakly positive and a desmin stain was strongly positive, especially in the more pleomorphic spindle cell areas (Fig. 6)
Diagnosis: “Chondrosarcoma, Osteosarcoma, Myosarcoma and Fibrosarcoma of the atrium (‘Malignant Mesenchymoma’)â€
Timothy R. Smith, MS3 and Donald R. Chase, MD
Department of Pathology and Human AnatomyÂ
Loma Linda University and Medical Center, Loma Linda, CaliforniaÂ
California Tumor Tissue Registry, Loma Linda, California
Discussion: Primary tumors of the heart are very rare. Â They are mostly benign with relative frequencies of:
myxomas (78%)
sarcomas of any type (10%)
fibroma (3%)
lipoma (3%)
vascular tumors (2%), andÂ
rhabdomyoma (2%). Â Â
Most cardiac tumors present with non-specific symptomology reflecting disruption of the conduction system or blockage of blood flow.  In addition, high grade sarcomas may also present with distant metastasis, especially when they involve the great vessels of the heart.  When malignant, cardiac tumors (virtually all of which are sarcomas) are usually of a “pure†histologic type and the combination of sarcoma subtypes in our presentation case is exceedingly rare.  Although it is common practice to identify the specific type of sarcoma by morphologic and immunohistochemical patterns, the exercise of doing so is of little clinical value.  In addition to location of a sarcoma in the great vessels, low resectibility, a high mitotic rate and the presence of necrosis portend an accelerated aggressive behavior.
There is still debate about the classification of soft tissue tumors that show more than one tumor type.  In soft tissue, the uterus, and other in other organs, when both an epithelial and sarcomatous component are present the term, “carcinosarcoma†is still in use.  In soft tissue the term, “malignant mesenchymoma†has been used since it was first coined by Stout in 1948.  He defined these complex neoplasms as “non-epithelial malignant tumors showing two or more unrelated tissue types in addition to a fibrosarcomatous element† Stout and Lattes later changed this definition to mean sarcomas composed of two or more unrelated tumor elements, with or without a fibrosarcomatous component. More recently, the World Health Organization decided that “malignant mesenchymoma†was a vague term and did not reveal the various morphologies of the mixed sarcomas.  The current thought, expounded by Weiss, Goldblum, and Fletcher is that the term “malignant mesenchymoma†should be deleted from the classification scheme of soft tissue tumors.Â
Differential Diagnosis of Cardiac Neoplasms:
• Cardiac Rhabdomyomas usually arise in the myocardium of children, generally below the age of five years and is sometimes associated with tuberous sclerosis. The cells are usually “bubblyâ€, stain for desmin, and may have striations.  Mitotic figures are virtually non-existent.  Occasional rhabdomyomas may spontaneous regress, usually by maturation into more normal-appearing myocytes.Â
Â
• Cardiac Fibromas also arise mostly in children and may be associated with adenoma sebaceum of the skin and kidney tumors.  They consist of a uniform proliferation of fibroblasts without significant mitotic activity.  They lack either a cartilaginous or an osseous matrix.
• Myxomas have varied gross appearances and are smooth or lobular, firm or gelatinous, and contain a myxoid stroma. They account for the majority of primary cardiac tumors. Seventy-five percent are found in left atria.  Curiously, benign myxomas usually are found in the left side of the heart, while sarcomas are usually found in the right side.
• Vascular neoplasms may be benign (hemangioma) or malignant (angiosarcoma) and when large, may interrupt blood flow through the heart.  Angiosarcoma is particularly prone to early metastases.  Both tumors stain for vascular markers such as CD31, CD34 and HFVIII.
• Lipomas usually originate in the endocardium or epicardium and have a broad pedunculated base.  They consist of mature, adult-type fat.
• Pericardial cysts may mimic a cardiac tumor or pericardial effusion on chest x-ray.  They are usually asymptomatic, although some cause compressive symptoms (eg, chest pain, dyspnea, cough).  The center of the process is of blood-tinged fluid which is bounded by pericardial-type cells.Â
• Sarcomas are usually vimentin-positive and are composed of spindled cells of varying pleomorphism.  A negative desmin stain tends to exclude myogenous origin and a negative S100 stain helps in the exclusion of neural tumors and melanoma.  A negative epithelial stain helps rule out spindled carcinomas and synovial sarcoma.  Osseous and/or cartilaginous matrix formation rules out the previously mentioned benign cardiac tumors.
• Teratomas usually occur in midline structures such as the anterior mediastinum but rarely arise in the heart.  By definition, these germ cell tumors contain tissue types representing all three germ layers.
A high index of suspicion is necessary for early diagnosis of these cardiac neoplasms. Â Patients with signs of cardiac obstruction, chest pain or discomfort, or dyspnea should have appropriate radiologic and laboratory work-ups. Â But even with early diagnosis, sarcomas do poorly, and even with positive indicators such as low mitotic rate, low levels of necrosis, presence in the left side of the heart, and good resectibility, the survival rate of cardiac sarcomas is under five years. Â Cardiac transplant is a last measure for patients with Unmanageable primary cardiac tumors.Â
Suggested Reading:
Weiss SW, Goldblum JR. Enzinger & Weiss’ Soft Tissue Tumors 5th ed: 1213-1214, 2008.
Stout AP. Mesenchymoma: the mixed tumor of mesenchymal derivatives. Annu Surg: 127:278-90, 1948.
Stout AP, Lattes R. Tumors of the soft tissues. In: Firminger GI, ed. Atlas of Tumor Pathology. 2nd series. Fascicle 1. Washington, DC: Armed Forces Institute of Pathology;: 172-5, 1967
Virmani, Burke, Farb, Atkinson. Tumors and tumor-like lesions of the heart and great vessels. Cardiovascular Pathology 2nd ed;: 424-458, 2001.
Merck online manual section cardiovascular disorders, subject Cardiac tumors, Topics Cardiac tumors, 2008.
 History: A 43-year-old gravida 0 woman with a history of menorrhagia was found to have a 3.5 cm left retroperitoneal, periaortic mass on abdominal CT six years previously (
History: A 43-year-old gravida 0 woman with a history of menorrhagia was found to have a 3.5 cm left retroperitoneal, periaortic mass on abdominal CT six years previously (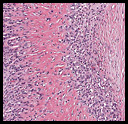 History: A 28 year-old woman presented with a palpable mass in the deep tissues of the left thigh. A 4.6 x 3.5 x 2.8 cm ovoid rubbery soft tissue mass was removed. Its perimeter was well-circumbscribed and was partially covered by gray-brown fragments of skeletal muscle. The cut surface was glistening and pink-white. Rare microcysts were seen.
History: A 28 year-old woman presented with a palpable mass in the deep tissues of the left thigh. A 4.6 x 3.5 x 2.8 cm ovoid rubbery soft tissue mass was removed. Its perimeter was well-circumbscribed and was partially covered by gray-brown fragments of skeletal muscle. The cut surface was glistening and pink-white. Rare microcysts were seen.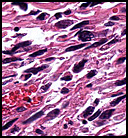 History: A 42-year-old woman with a one month history of dry cough was radiographically thought to have a left cardiac myxoma and bilateral hydrothorax. At surgery, however, she was found to have a 7.0 x 6.0 x 4.5 cm gray fleshy solid mass in the left pulmonary vein which extended into the left atrium.
History: A 42-year-old woman with a one month history of dry cough was radiographically thought to have a left cardiac myxoma and bilateral hydrothorax. At surgery, however, she was found to have a 7.0 x 6.0 x 4.5 cm gray fleshy solid mass in the left pulmonary vein which extended into the left atrium. History: A 20-year-old woman presented with chronic jaw pain and swelling in the left cheek. Computerized tomography (CT) scan identified an expansile, multilocular, osteolytic mass in the left lower maxillary sinus (
History: A 20-year-old woman presented with chronic jaw pain and swelling in the left cheek. Computerized tomography (CT) scan identified an expansile, multilocular, osteolytic mass in the left lower maxillary sinus (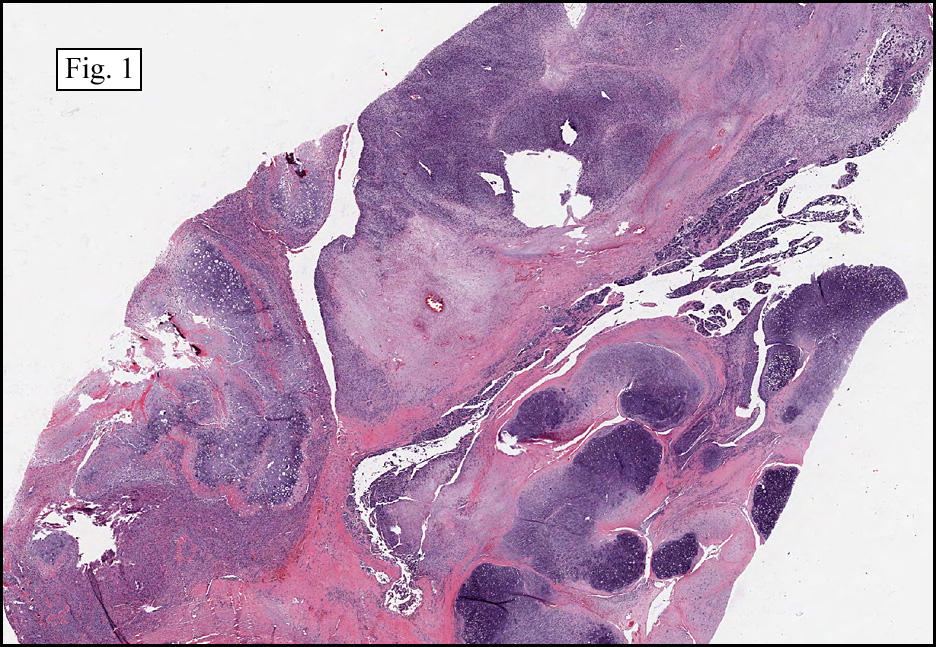{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}