History: A 76 year old man with a one year history of progressive right-sided hearing loss and tinnitus presented with a four month history of frequent falls. A physical examination was essentially normal. The tympanic membranes were clear, without drainage. An MRI of the head revealed a 1.2 x 1.1 x 1.0 cm peripherally-enhancing lobulated mass involving the right internal auditory canal with extension into the right cerebellopontine angle. The clinical impression was schwannoma vs. meningioma.
At surgery, two 0.2 cm fragments of red-tan tissue were removed from the right cerebellopontine angle. They exuded “cheesy-white†material.
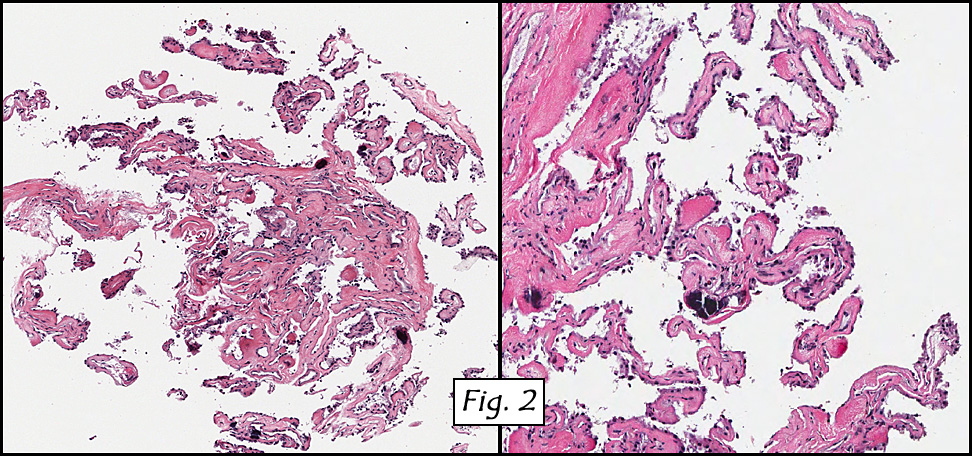
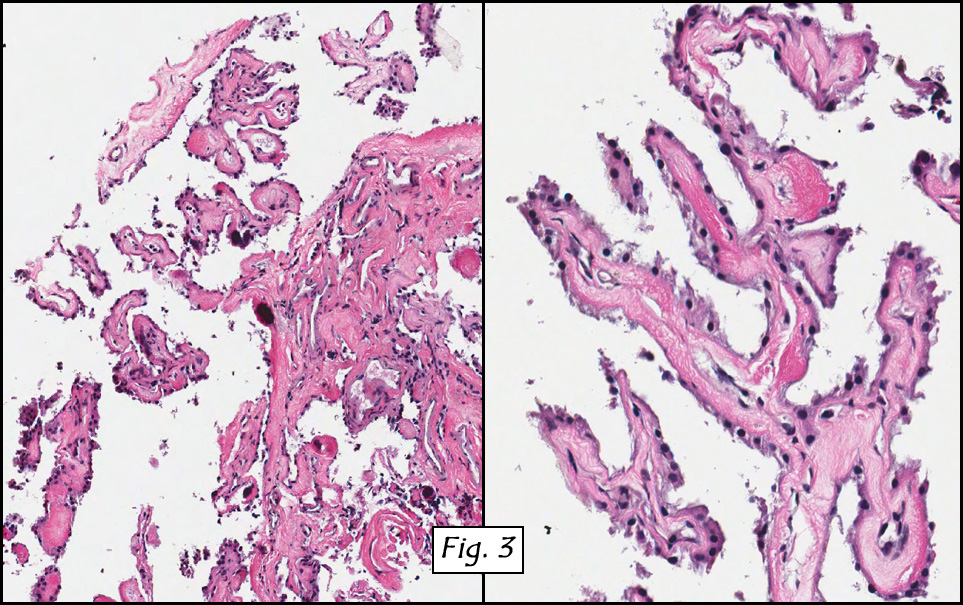
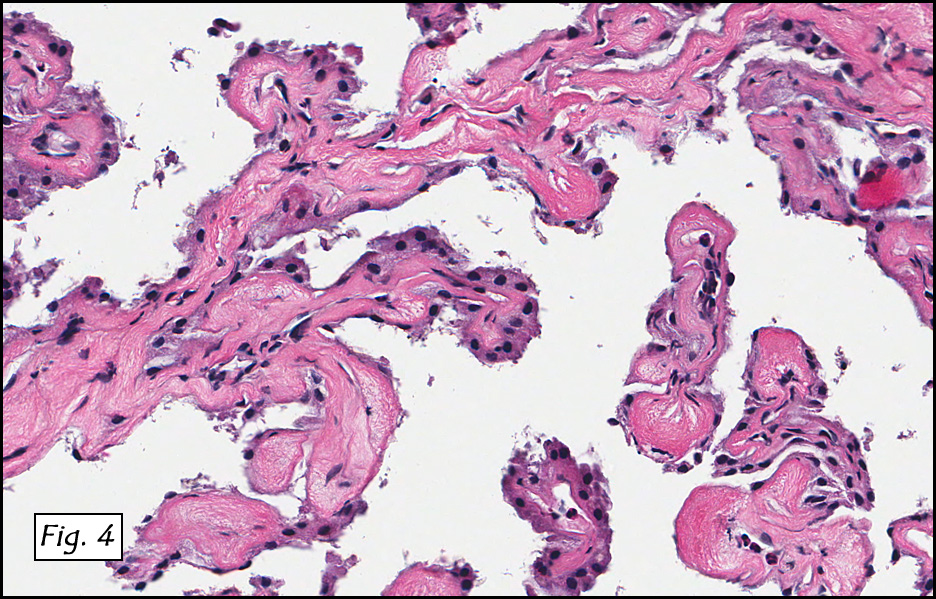
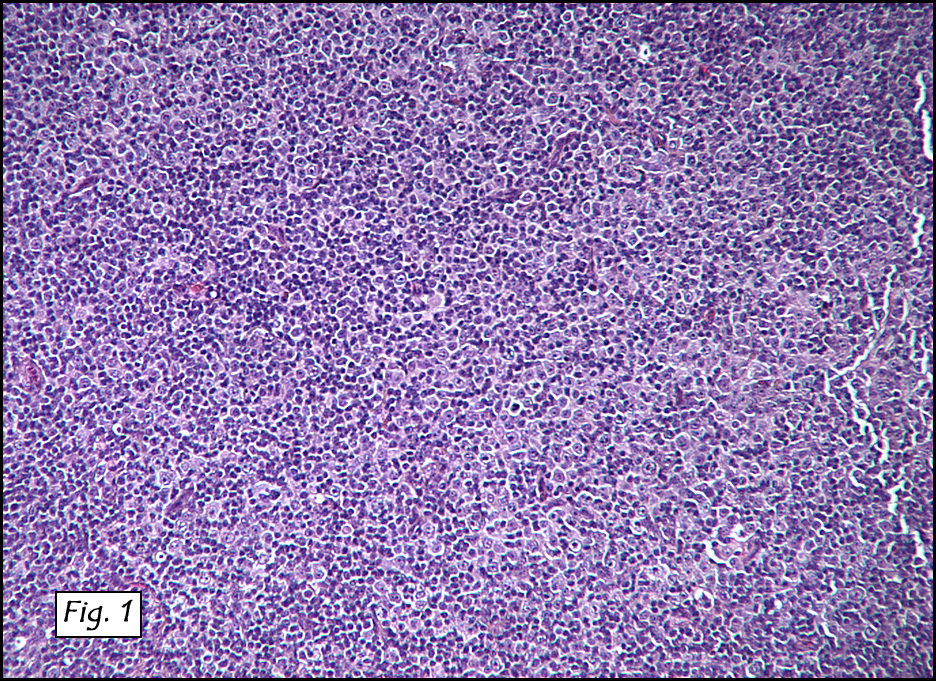
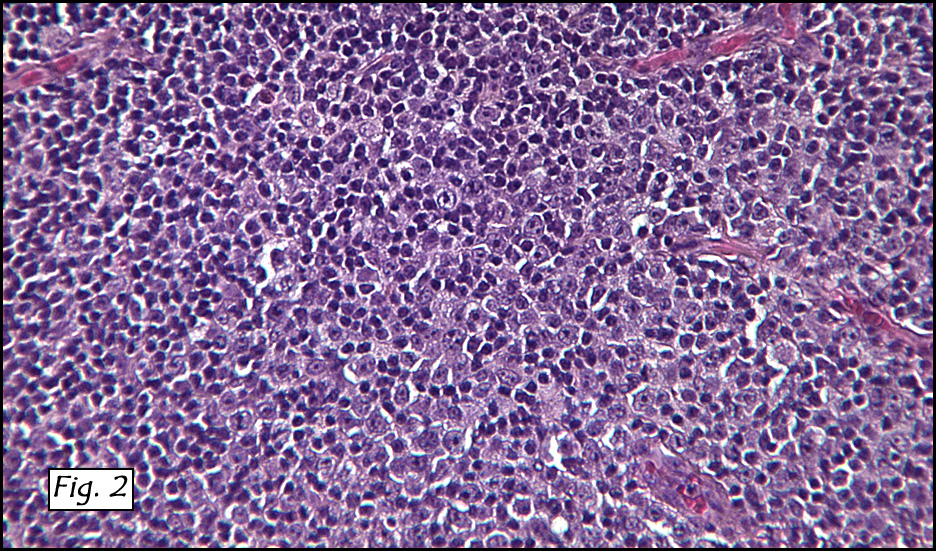
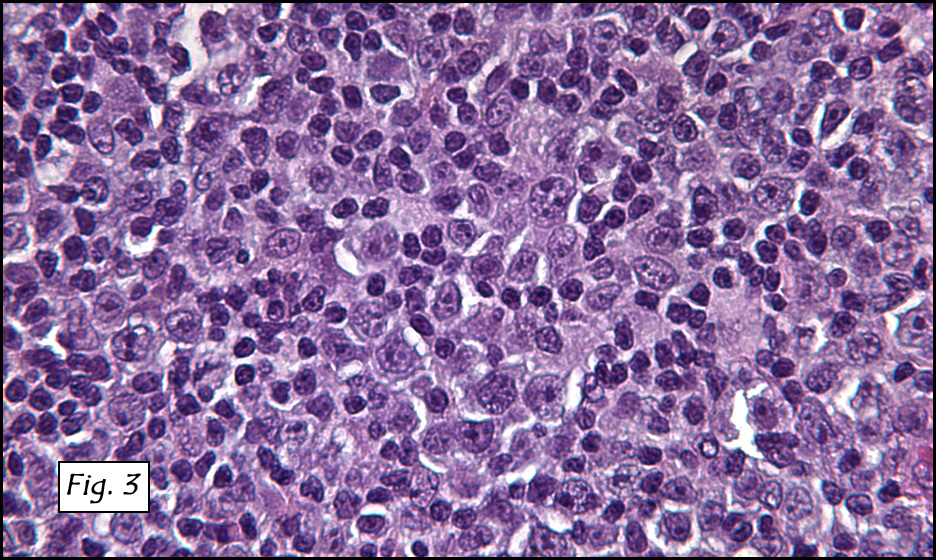
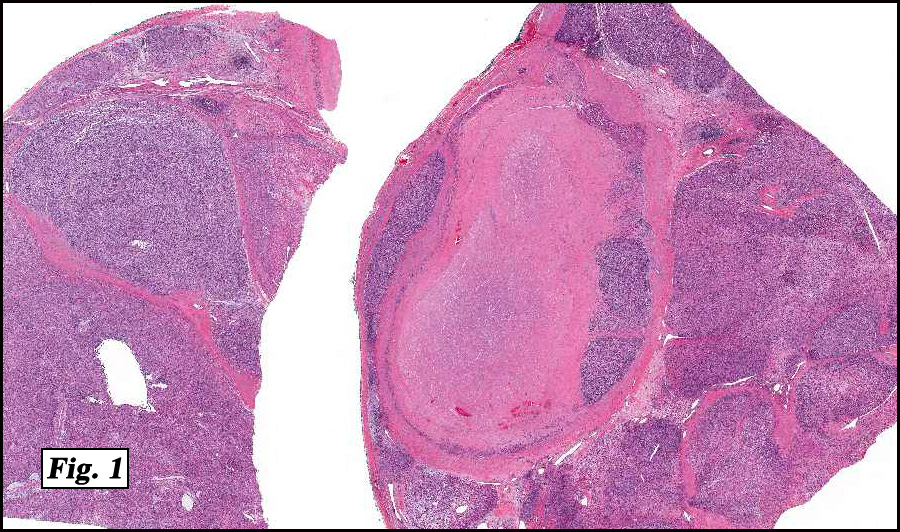
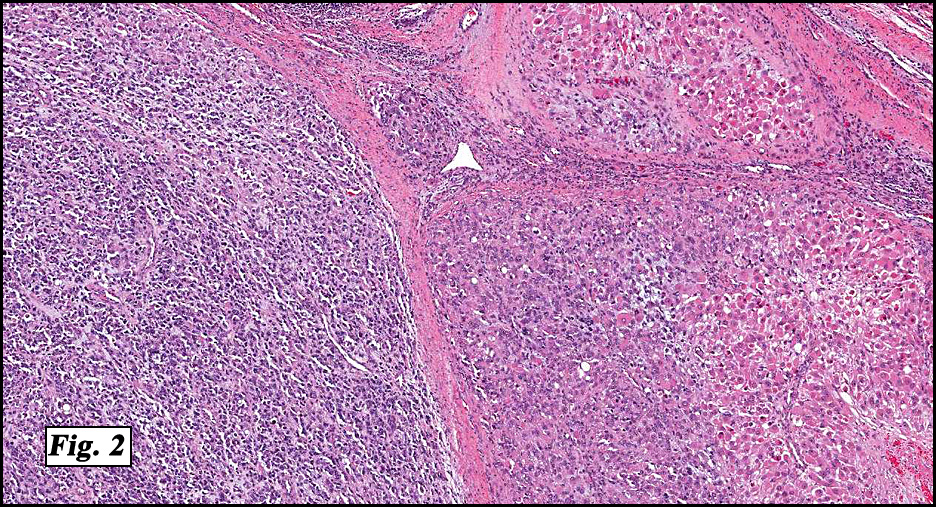
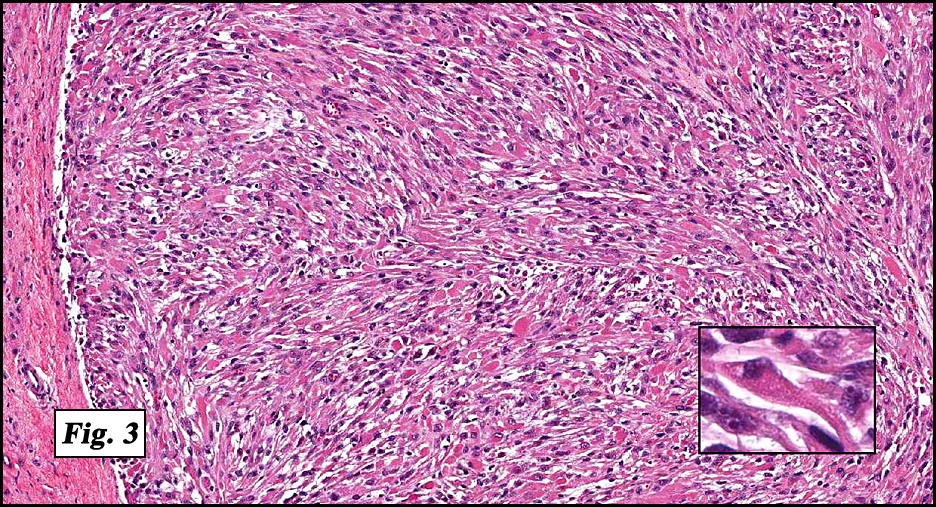
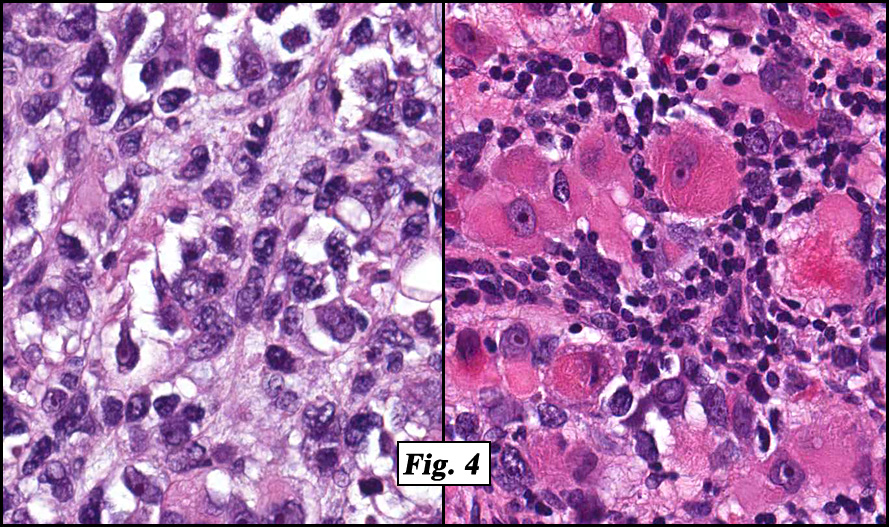
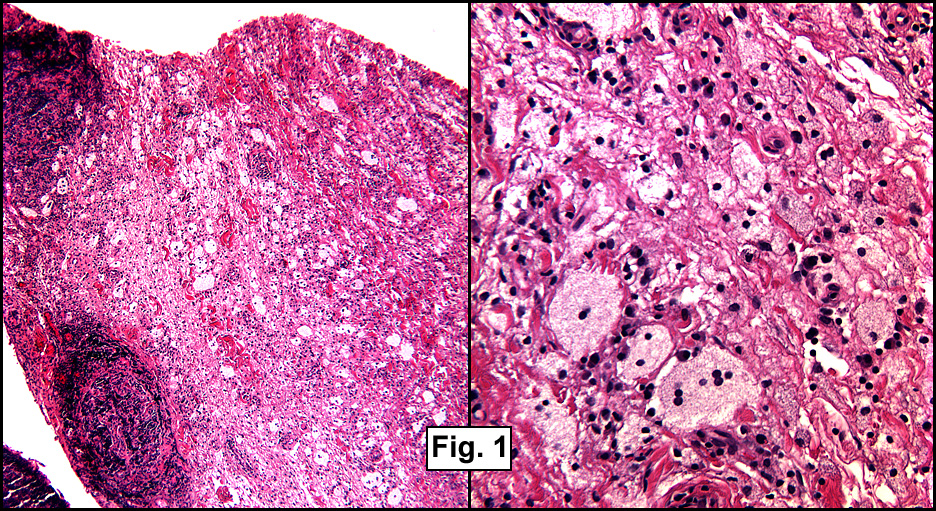
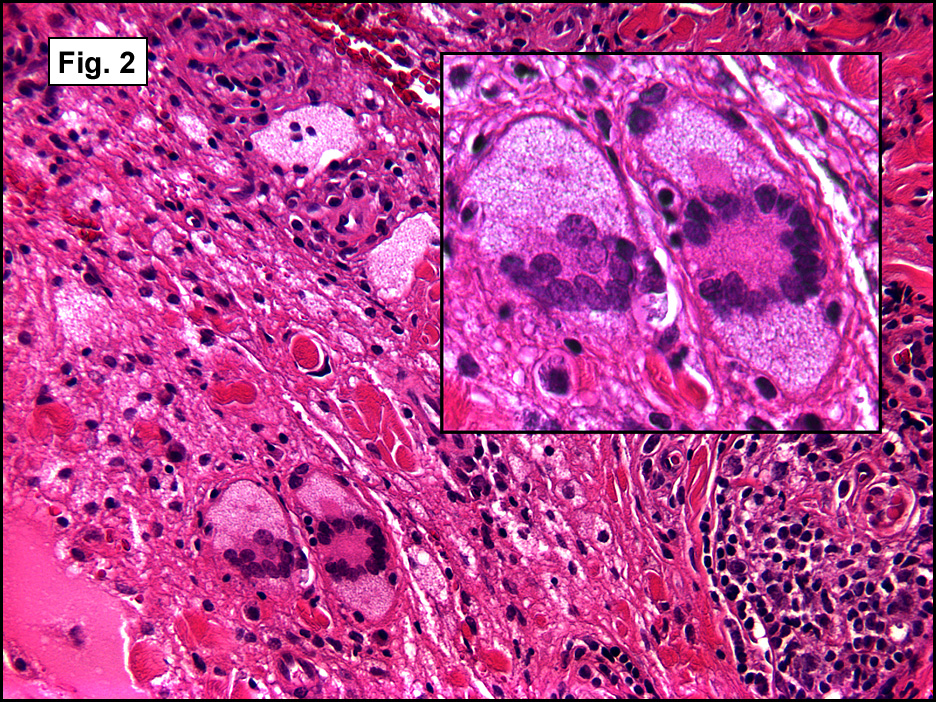
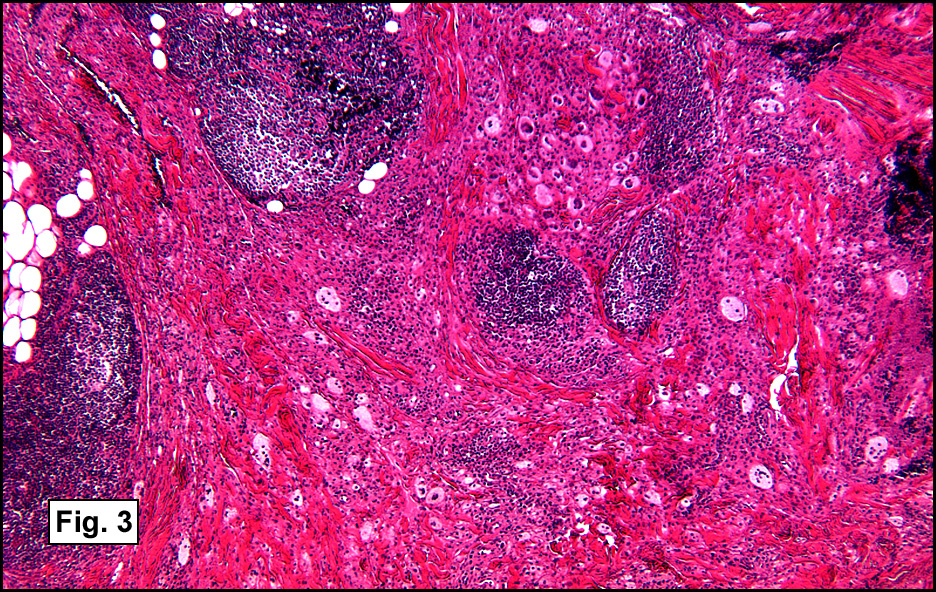
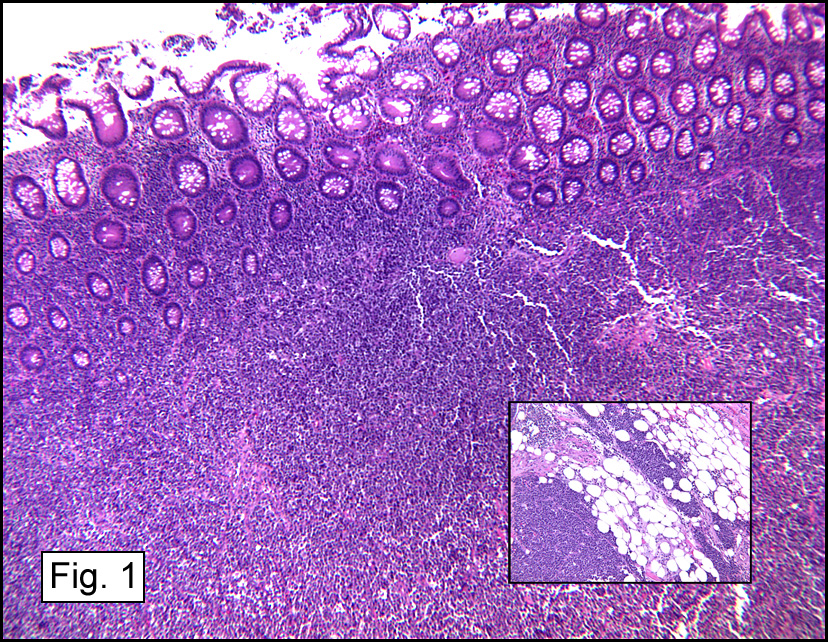
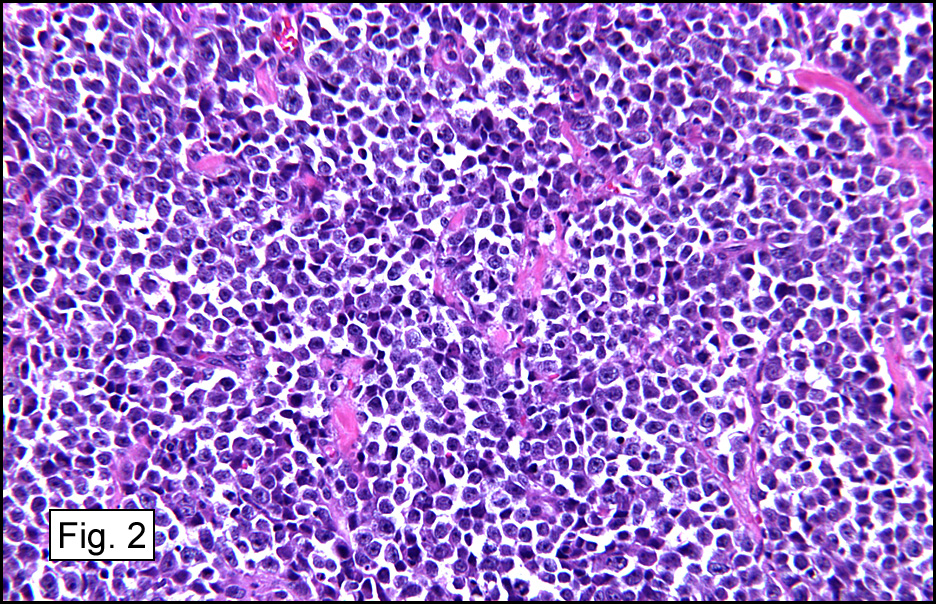
Microscopic sections revealed a papillary tumor (Fig. 1) with spaces lined by a single layer of bland cuboidal cells with acidophilic cytoplasm. The supporting stroma was fibrovascular and showed focal calcifications and perivascular hyalinization (Figs. 2,3,4). Neither significant pleomorphism nor mitotic activity was seen.
Diagnosis: “Low grade adenocarcinoma of endolymphatic sac origin (Heffner tumor)â€
Hannah Wong MD, Norman Peckham MD, Donald Chase MD
Department of Pathology, Loma Linda University and Medical Center, Loma
Linda, California
California Tumor Tissue Registry, Loma Linda, California
Discussion: Low grade adenocarcinoma of endolymphatic sac origin (LGAE) is also referred to as endolymphatic sac papillary tumor, aggressive papillary tumor of temporal bone, endolymphatic sac adenoma, temporal-mastoid bone adenoma/adenocarcinoma, low grade adenocarcinoma of potential endolymphatic sac origin, and aggressive papillary tumor of the temporal bone.
LGAE is a rare tumor first described by Heffner in 1989 as an aggressive neoplasm arising in the region of the temporal bone. There is no apparent gender predilection. Of curiosity is that 11-30% of patients with LGAE also have von Hippel-Lindau (VHL) disease. The average age of patients with LGAE (alone) is 52.5 years, but is younger (31.3 years) when associated with VHL disease. Most tumors are unilateral, but patients with VHL have a higher incidence of bilateral disease.
Clinically, patients may present with sensorineural hearing loss, tinnitus, vertigo, serous otitis media, cranial nerve paralysis and/or jugular foramen syndrome. The tumor may be found on physical examination as a blue or red mass behind an intact or ruptured tympanic membrane. Imaging studies generally show soft tissue involvement with varying degrees of local destruction usually at the posterior aspect of the petrous portion of the temporal bone. More advanced tumors may show intraosseous extension.
Histologically, the tumor is predominantly papillary with a variable degree of cystic change. The papillae have fibrovascular cores lined by a single layer of cuboidal to columnar cells. They may have a follicular appearance or be “crowded†into a solid mass. Individual cells may show clear, vacuolated or acidophilic cytoplasmic features and usually have distinct cell borders. The nuclei are oval, mildly irregular, centrally to apically located and have inconspicuous nucleoli. Mitotic activity is generally absent. The surrounding stroma is hypocellular with focal areas of fibrosis, hemorrhage, and/or cholesterol clefting. PAS-positive proteinaceous material similar to colloid may be present.
LGAE generally displays one or both of two main histological patterns:
•   Follicular pattern consisting of colloid filled follicles.  This pattern is believed to be secondary to cystic dilation of the glands, resulting in a follicle-like appearance similar to that seen in thyroid tumors.
•   Papillary pattern consisting of a more cellular lesion made of papillary and solid areas.
Although histogenesis is still debated, most studies suggest that LGAE originates from the endolymphatic sac, which in turn was derived from neuroectoderm (otocyst) between the dura and the posterior surface of the petrous portion of the temporal bone. The tumor strongly resembles normal endolymphatic sac tissue consisting of papillary epithelium composed of cuboidal or columnar cells arranged in villous folds overlying loose connective tissue.
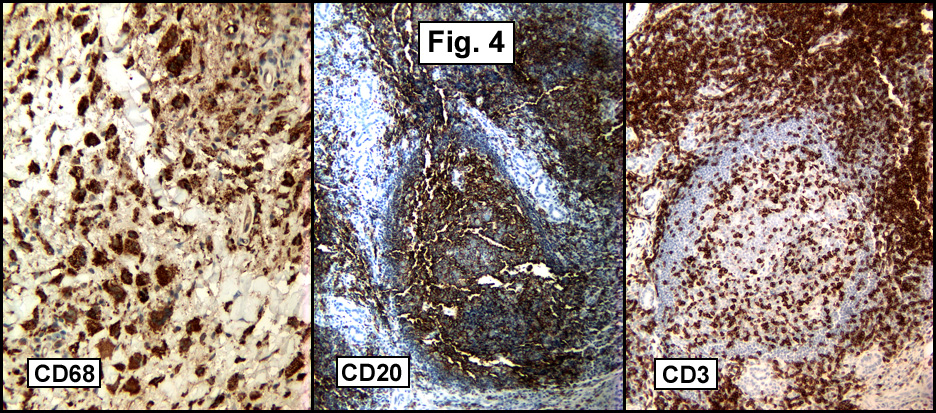
LGAE expresses several cytokeratins including CAM5.2, 34βE-12, CK7, CK8 and CK19. It also usually marks for epithelial membrane antigen and vimentin. Expression of vascular endothelial growth factor and neuron specific enolase may also occur and increased Ki-67 expression has been reported.
The differential diagnoses for LGAE includes middle ear adenoma, middle ear carcinoma, paraganglioma, meningioma, squamous cell carcinoma and primary and secondary bone lesions. Of these lesions, middle ear adenoma is the most common tumor with which LGAE can be confused with. Middle ear adenomas are less aggressive neoplasms than LGAE and are typically confined to the middle ear. Unlike LGAE they usually show glandular-trabecular growth patterns.
Despite local aggressiveness, clinical progression of LGAE is generally slow, allowing for initial local surgical resection without use of chemotherapy or radiotherapy. Postoperative radiotherapy is generally reserved for recurrent disease or for cases of persistent disease due to previous incomplete excision.
Suggested reading:
1.   Mills SE, Faggey MJ and HF Frierson, Jr. Aggressive papillary tumor of temporal bone and endolymphatic sac (low-grade adenocarcinoma of endolymphatic sac origin). Tumors of the upper aerodigestive tract and ear. AFIP. Third series, fascicle 26; 436-438.
2.   Horiguchi H, Sano T, Toi H, Kageji T, Hirokawa M, Nagahiro S. Endolymphatic sac tumor associated with a von Hippel-Lindau disease patient: an immunohistochemical study. Mod Pathol. 14(7):727-732, 2001.
3.   Yilmaz I, Bolat F, Demirhan B, Aydin V, Ozluoglu LN. Endolymphatic sac papillary tumor: a case report and review. Auris Nasus Larynx. 35:276-281, 2007.
4.   Delisle MB, Uro E, Rouquette I, Yardeni E, Rumeau JL. Papillary neoplasm of the endolymphatic sac in a patient with von Hippel-Lindau disease. J Clin Pathol. 47:959-961, 1994.
5.   Ho VT, Rao VM, Doan HT, Mikaelian DO. Low-grade adenocarcinoma of probable endolymphatic sac origin: CT and MR appearance. AJNR. 17:168-170, 1995.
6.   Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin: a clinicopathologic study of 20 cases. Cancer. 64(11):2292-2302, 1989.
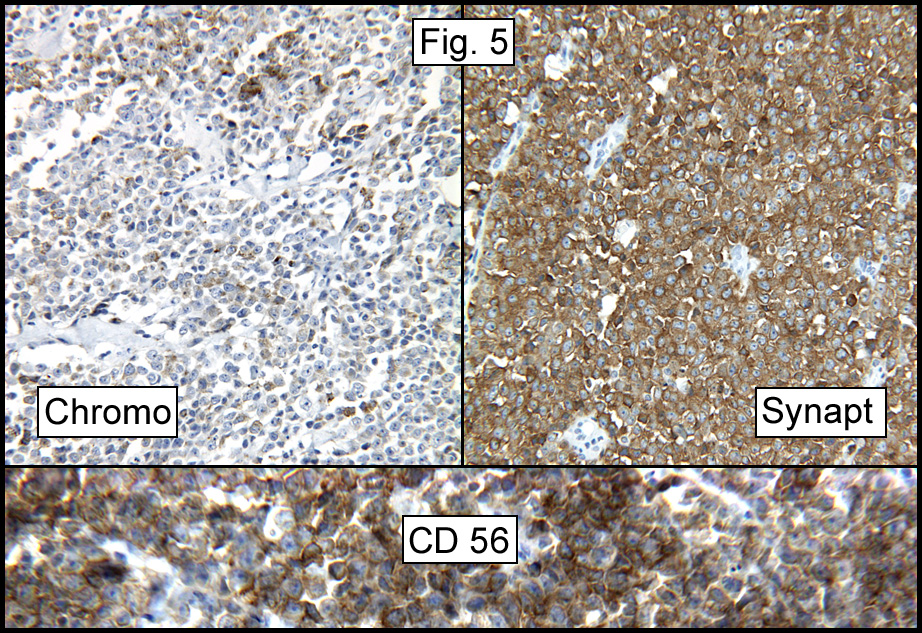
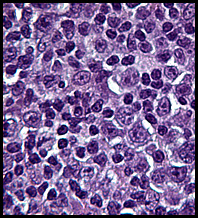 History: A previously healthy 80 year-old man presented with a one-month history of right axillary lymphadenopathy. Physical examination revealed a mildly tender, firm, two cm lymph node in the right axilla. No other lesions identified.
History: A previously healthy 80 year-old man presented with a one-month history of right axillary lymphadenopathy. Physical examination revealed a mildly tender, firm, two cm lymph node in the right axilla. No other lesions identified.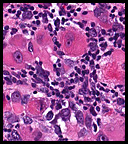 History: An 8 year old otherwise healthy boy was found to have a left scrotal mass. An ultrasound showed the left testis to be “inhomogeneous†and surrounded by a tumor confined to the scrotum. The right testis was normal. Regional inguinal lymph nodes were not enlarged and an abdominal CT scan was unremarkable.
History: An 8 year old otherwise healthy boy was found to have a left scrotal mass. An ultrasound showed the left testis to be “inhomogeneous†and surrounded by a tumor confined to the scrotum. The right testis was normal. Regional inguinal lymph nodes were not enlarged and an abdominal CT scan was unremarkable.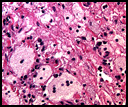 History: A 55-year-old man with a ten year history of swollen eyes presented with asthmatic symptoms. He was found to have enlarged upper and lower eyelids with ocular secretions that prevented him from seeing properly. The clinical differential included inflammatory pseudotumor of the orbits, proptosis from Grave’s disease and multiple myeloma. The workup showed an elevation of free kappa light chains. A periocular biopsy was performed.
History: A 55-year-old man with a ten year history of swollen eyes presented with asthmatic symptoms. He was found to have enlarged upper and lower eyelids with ocular secretions that prevented him from seeing properly. The clinical differential included inflammatory pseudotumor of the orbits, proptosis from Grave’s disease and multiple myeloma. The workup showed an elevation of free kappa light chains. A periocular biopsy was performed.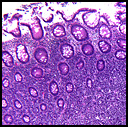 History: A 53 year old woman presented with acute abdominal pain assumed to be diverticulitis. A right hemicolectomy was performed.
History: A 53 year old woman presented with acute abdominal pain assumed to be diverticulitis. A right hemicolectomy was performed.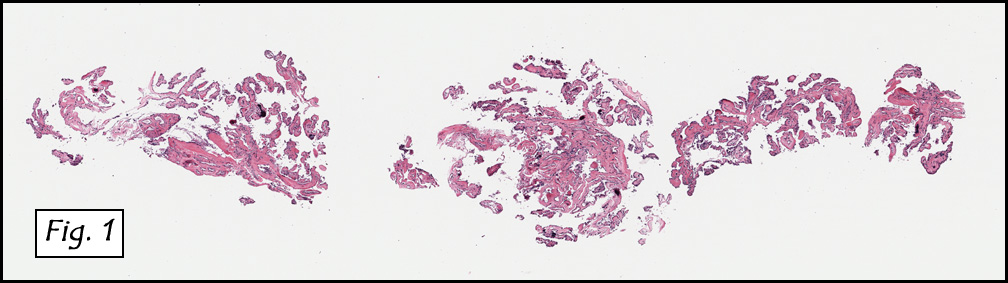{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}